
Research
Exploring protein systems across biological scales
I utilize methods that allow me to explore bacterial sensory systems across measurable scales. Here, I will explain these methods and how they can answer questions starting at the cell behavioural level and all the way down to the structure/function of single proteins.
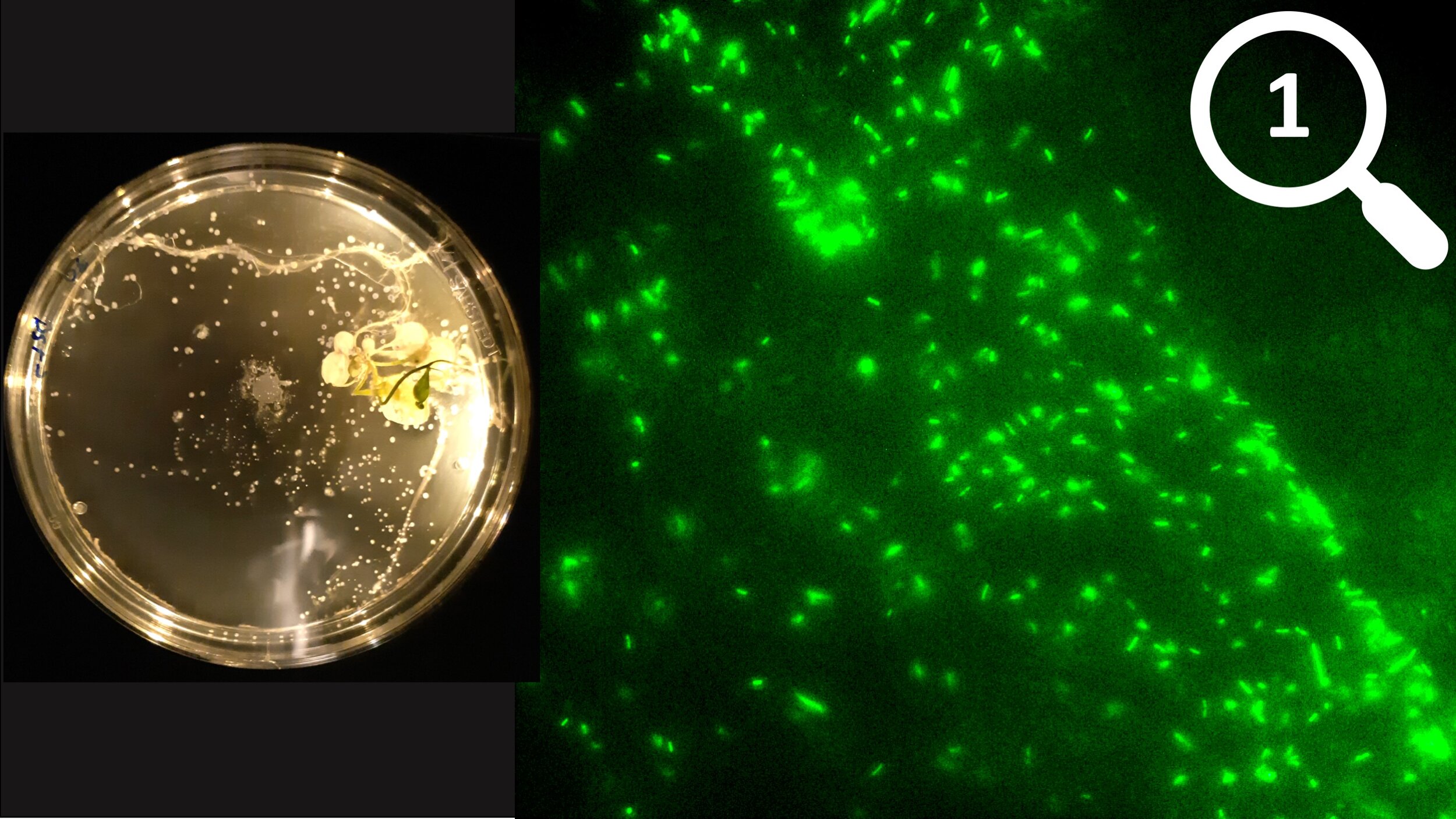
Microbial motility
At the largest scale, I explore microbial behaviour in response to their environment. Specifically, I look at microbial motility to determine how microbes sense and respond to their environment. For instance, I can see through motility assays on agar plates that Streptomyces spores preferentially germinate near plant tissues, or use fluorescence microscopy to see how motile bacteria move toward a nutrient source and form biofilms. However, these methods do not give structural insights at the sub-cellular level.

Cell Phenotypes
To see cellular features, I use cryo-electron microscopy (cryo-EM). This method allows me to see what cells look like and what structures they have. For example, I can see that the pathogenic Spirochete Treponema denicola is a long, tube-shaped organism with many flagella that wrap around the cell body. I can also see a chemotaxis array near the cell pole (red box). Although I can see the chemotaxis array within cryo-EM images, I cannot determine what the chemotaxis apparatus looks like.

Sub-cellular structures
I can elucidate the structures of cellular complexes by conducting cryo-electron tomography (cryo-ET) followed by sub-tomogram averaging. Essentially, cryo-ET is conducted by taking cryo-EM images of a sample at several different angles. Using these images, I can reconstruct what the sample looks like in 3D. Then, I can select my cellular complexes of interest and average thousands of them to get a single high-resolution structure of complex. As an example, I am showing the structure of the chemotaxis array in Treponema denticola. The sub-tomogram averages show protein densities (black) of the chemotaxis array. Now, I can design a structural model of the array. As mentioned, I could not get this information from cryo-EM alone.

All-atom protein structures
With cryo-ET I can see the densities of proteins inside intact, native cells. However, this method is rarely able to generate densities with high enough resolution to determine the complete all-atom model. To answer such questions, I use x-ray crystallography of isolated proteins. In summary, I purify proteins from cells and then force them to form highly uniform crystals. Typically, this method allows structure determination down to 1 Angstrom. Here, I’m showing the 1.5 Angstrom crystal structure of a Treponema denticola chemotaxis protein (PDB ID: 6Y1Y). From cryo-ET, I was able to see density corresponding to this protein but I was unsure of what it looked like. Now I can see that it is a helical coiled-coil dimer that is interrupted by two flexible loops.

Protein mechanisms and dynamics
So far, I’ve shown you the techniques I use that let me investigate proteins all the way down to all-atom structures. But the exploration doesn’t stop there! Cryo-ET and crystallography are powerful but only give you static images of molecules. In cells, proteins can be highly dynamic so a lot of information can still be missing on a structural level. Furthermore, enzymes can have complex mechanisms that involve residue modifications and/or the involvement of ligands and cofactors. To determine how proteins behave, I use a variety of structural, biochemical, and spectroscopic methods. Such studies have allowed me to ascertain the oxygen-binding cycle of the oxygen sensor ODP, and large-scale structural rearrangements of the chemotaxis protein CheA.